By CHAD TERHUNE
For Kaiser Health News
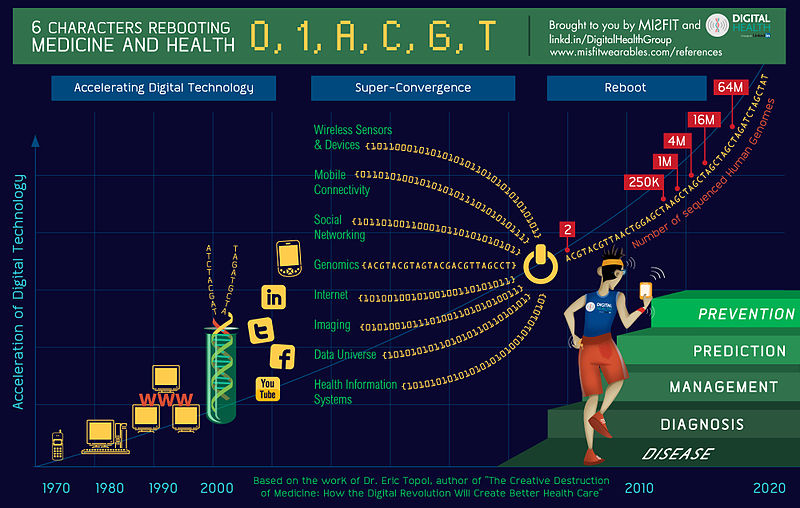
Federal regulators said 12 U.S. hospitals, including well-known medical centers in Los Angeles, Boston, and New York, failed to promptly report patient deaths or injuries linked to medical devices.
The Food and Drug Administration publicly disclosed the violations in inspection reports this week amid growing scrutiny of its ability to identify device-related dangers and protect patients from harm.
Some of the reporting lapses were found at Massachusetts General Hospital, in Boston, NewYork-Presbyterian Hospital and two hospitals in Los Angeles — the Ronald Reagan UCLA Medical Center and Cedars-Sinai Medical Center.
Dr. Jeffrey Shuren, director of the FDA’s Center for Devices and Radiological Health, said the violations pointed to a larger problem among hospitals nationwide in reporting patient harm tied to medical devices.
“We believe that these hospitals are not unique in that there is limited to no reporting to FDA or to the manufacturers at some hospitals,” Shuren wrote in an agency blog post this week. “Hospital staff often were not aware of, nor trained to comply with, all of the FDA’s medical device reporting requirements.”
Under federal rules, hospitals must within 10 days report serious injuries potentially caused by devices to the manufacturer and notify both the manufacturer and the FDA about any deaths that may have resulted. Manufacturers are required to file reports to the FDA within 30 days of learning about an injury or death that may have been caused by a device.
Among the 17 hospitals reviewed, the FDA said six didn’t properly report both patient deaths and injuries linked to devices within 10 days as required. Five other hospitals didn’t report serious injuries in a timely manner, according to the FDA. The inspection at one hospital, NewYork-Presbyterian, focused only on a death.
NewYork-Presbyterian said it filed medical device reports “in accordance with FDA regulations” and none of the agency’s findings related to the quality or safety of patient care.
It’s hard to discern what devices were involved or other details in many of these cases because the inspection reports are brief and partly redacted by the FDA. The inspection reports indicate that in some cases hospitals reported events late and in others not at all.
At Massachusetts General, an FDA investigator found reporting delays of 10 months and 18 months in two separate patient deaths related to devices.
In a statement, hospital spokeswoman Terri Ogan said the FDA’s findings all have been addressed. “Massachusetts General Hospital takes its reporting obligations very seriously and strives to comply with all requirements in a comprehensive and timely manner,” she said.
At Huntington Memorial Hospital, in Pasadena, Calif., an FDA investigator found that a patient died from complications related to a multi-drug resistant infection and cardiac arrest following a procedure involving a duodenoscope, a long and flexible instrument put down a patient’s throat.
According to FDA records, the hospital learned through test results that the patient’s infection was likely related to 14 other confirmed infections caused by contaminated duodenoscopes. “However, this death was not reported to the FDA and the manufacturer by your facility,” the FDA inspector wrote in a December 2015 report.
A spokeswoman for Huntington Memorial, Eileen Neuwirth, said “we have taken steps to ensure rigorous compliance going forward.”
The FDA findings underscore concerns raised by a U.S. Senate report in January, which exposed reporting failures by hospitals as well as mistakes by device makers that contributed to multiple superbug outbreaks across the U.S. from contaminated duodenoscopes. The FDA’s oversight of medical devices was also faulted in the report.
As many as 350 patients at 41 medical centers worldwide have been infected or exposed to contaminated duodenoscopes from 2010 to 2015, according to the FDA.
The agency initiated its investigation of hospitals’ reporting in December 2015, a month before the Senate report was released. But the agency was already under fire by then for spotty oversight of duodenoscope manufacturers and other devices.
Shuren said in his blog post that the agency focused on hospitals where safety issues had occurred involving either duodenoscopes or power morcellators, a surgical tool used in hysterectomies. Morcellators are used to cut up benign growths called fibroids, but the FDA has warned about the device spreading cancerous tissue in the abdomen and pelvis. The investigators examined incidents involving other devices as well.
Other than publicly announcing the violations, Shuren said the agency didn’t plan on taking further action against the hospitals. Instead, he said he wants to work with the hospital industry to improve monitoring of devices.
“We feel certain there is a better way to work with hospitals to get the real-world information we need, and we should work with the hospital community to find that right path,” Shuren wrote.
Lawmakers, health policy experts and the FDA have proposed various reforms aimed at strengthening device surveillance, including tracking insurance claims data to supplement the injury reports and automating “adverse event” reports through electronic health records.
The issue may take on more urgency after federal authorities this month highlighted the infection risk from yet another commonly used device — heater-cooler units used in open-heart surgeries. The FDA is holding a public meeting Dec. 5 on improving hospital-based surveillance of devices.
According to the FDA, the hospitals that didn’t report deaths as required were Advocate Lutheran General, in Park Ridge, Ill.; Huntington Memorial Hospital; Reading Hospital and Medical Center, in West Reading, Pa.; Allegheny General Hospital, in Pittsburgh; NewYork-Presbyterian; and two in Boston — Brigham and Women’s Hospital and Massachusetts General.
The agency said those that failed to report serious injuries in time were UCLA; Cedars-Sinai; Virginia Mason Medical Center. in Seattle; UMass Memorial Medical Center. in Worcester, Mass.; and Dartmouth-Hitchcock Medical Center, in Lebanon, N.H.
The FDA inspection for Advocate Lutheran General Hospital refers to 10 deaths related to a scope-related outbreak of carbapenem-resistant enterobacteriaceae, a superbug known as CRE. But a spokeswoman for the hospital said a “review of medical records in all 10 cases confirmed the cause of death was not linked to CRE.”
Dr. Leo Kelly, vice president of medical management at Advocate Lutheran, said \the hospital will continue to work with the FDA and manufacturers “to ensure the safety and well-being of our patients.”
Many of the hospitals involved said they welcomed the agency’s feedback and supported efforts to improve device oversight.
Cedars-Sinai said the FDA’s findings related to its use of a surgical stapler in June 2015.
UCLA said it promptly reported scope-related cases to the FDA but the agency asked for duplicate reports through a separate system.
Suzanne Anderson, president of Virginia Mason Medical Center, said the FDA’s recommendations on device reporting “will ultimately enhance patient safety across the country.”